|
|
Validation
We present here the results obtained using
the ConSeq server with 5 protein families: the C2 domain, the SH2 domain,
the SH3 domain, Pyruvate Kinase (PK) and HIV1-Reverse Transcriptase (HIV1-RT).
Their 3D structures exist, and precise annotation identifying the functional residues is available. Two different functional sites were identified in the SH2 domain, PK and HIV1-RT.
We run ConSeq
with external MSAs from the Pfam database (Bateman et al., 2002) for HIV1-RT
and PK and an MSA from the HOMSTRAD database (Mizuguchi et al., 1998) for the
SH3 domain.� For the C2 and the SH2 domains, the MSAs were generated
automatically using a query sequence (see Methodology). For each active site,
we calculated the overall success in identifying the functional residues and
analyzed the error contribution of Rate4Site and the surface accessibility
neural network algorithm.
The average success in
all the functional sites amongst the 5 proteins that were tested is 56%. Thus,
ConSeq is able to identify about half of the annotated functional residues. The
performance of the two algorithms independently, Rate4Site and the neural network, is 88% and
68%, respectively. The neural network algorithm error is caused by an overall 32% false prediction of exposed residues being buried in most cases ( except for the C2 domain and SH3 interface site in the
SH2 domain) and an 8% false prediction of buried residues being exposed instead of buried (in the active sites of PK and the C2 domain).
Rate4Site failed to
identify 12% of the functional residues as highly conserved (8 and 9 color
grade) in 3 cases: in HIV1-RT, Rate4Site failed to identify 3 residues as
highly conserved at the DNA/RNA binding site. In the SH2 domain, 2 residues in
the peptide binding site and 1 residue at the interface site with the SH3
domain were not identified. However, the conservation grades for these 6
residues are 6 and 7. Thus, these residues are assigned conservation grades above
the average ( Table 1).
It is important to keep in mind that the way we distinguish between the functional and structural residues ("functional" = highly conserved and exposed, whereas "structural" = highly conserved and buried) might be problematic in cases where a certain residue has both a functional role in e.g., ligand binding and catalysis, and a structural role in e.g., maintaining the conformation of the active site. Catalytic residues are often buried within the protein core due to their special enzymatic activity, and consequently could be identified inaccurately as structural rather than functional residues. Therefore, in some cases a strict classification of "functional" or "structural" residues could be imprecise (see the C2 domain example below). Thus it is always advisable to look first at the conservation grades; the suggestions regarding functional or structural residues should be taken 'with a grain of salt'.
Analysis
of ConSeq results
We first calculated the solvent-accessible surface area of the amino acids composing the 5 proteins and domains that were analyzed. Solvent-accessibility values were computed using the SurfV program with a probe radius of 1.4Å (Sridharan et al., 1992). To determine the relative surface accessibility r(i) of each residue i in the protein, we calculated r(i) = 100*acc(i)/max(i), where acc(i) is the solvent accessibility of residuei, as computed by the SurfV program (in Å2), and max(i) is the maximal accessibility of amino acid type i within the context of the tripeptide Gly-i-Gly. We chose a relative solvent accessibility threshold of 5% to discriminate between buried and exposed residues; thus residues with solvent accessibility beyond 5% of their maximum were assumed to be solvent exposed.
We defined as " functional", residues that have been reported in the literature to be involved in ligand binding, interaction sites etc. and are exposed to the solvent based on the threshold above.
The next step was to analyze the performance of ConSeq to predict the functional residues described above as highly conserved (color-coded conservation grades of 8-9) and exposed to solvent.
We calculated the performances of the Rate4Site and the neural-network algorithms both separately, and jointly.
For example, the 5 active site residues in HIV1-reverse transcriptase are: D110, Y183, M184, D185 and D186, with the last three directly involved in the catalysis (Ren et al., 1995). Running the SurfV program on HIV-1 RT structure (PDB: 1c1b) reveals that all 5 annotated residues are solvent-exposed. We carried out a ConSeq run using an external MSA of rvt domain from the Pfam database, with a retrovirus query sequence (SWISS_PROT: POL_HV1H2). The output points out that, according to the neural network algorithm 3 of the 5 residues are predicted to be solvent-exposed, while Y183 and M184 are predicted to be buried; i.e. the performance of the neural-net algorithm alone is 3/5*100 = 60%.
Regarding the conservation, all 5 residues are assigned as highly conserved: 8-9 conservation color grades. Therefore, Rate4Site performance alone is 5/5*100=100%.
When joining the performance of the two methods to detect the 5 annotated residues as highly conserved and exposed and consequently to be identified as " functional" (marked with " f"), only 3 of the 5 residues match these two conditions; i.e. the overall success is 3/5*100 = 60% for this active site.
False-negative predictions might influence the discrimination between the structural and functional predicted residues. In this example M184 is wrongly identified as structurally important, because it is predicted as highly conserved (9 color-grade) and buried.
The neural network's overall success for the 8 functional sites in detecting the exposed annotated functional residues as exposed is 34/50*100 = 68%. Rate4Site's overall success in detecting the exposed annotated functional residues as highly conserved is 44/50*100 = 88%, and the average performance of the two algorithms is 56% for the 8 functional sites.
A detailed summary of the results is provided in table 1 and in the diagram below:
Click on the protein or
domain name to view ConSeq results on-line
|
Protein/Domain
|
Functional
|
Exposed
|
ConSeq server
prediction
|
|
|
Exposed
|
Conserved
|
Exposed
residues
|
|
Notes
|
|
|
residues
|
residues
|
Residues
|
residues
|
(NeuralNet)
and
|
Success
|
|
|
|
(by
annotation)
|
(surfV5%)
|
Neural
Net
|
Rate4Site
(8+9 color grades)
|
conserved
(8+9)
|
%
|
|
|
SH2
domain
|
|
|
|
|
|
|
|
|
Peptide binding site
|
9
|
9
|
7
|
7
|
5
|
56%
|
|
|
SH3 interface
|
3
|
3
|
3
|
2
|
2
|
67%
|
|
|
Pyruvate
Kinase
|
|
|
|
|
|
|
|
|
Active site (pyruvate, K+, Mn2+, ATP)
|
11
|
8
|
4
|
8
|
4
|
50%
|
3 false positives predictions (exposed residue
Instead of buried)
|
|
FBP
binding site (allosteric regulation)
|
4
|
3
|
1
|
3
|
1
|
33%
|
|
|
C2
domain
|
|
|
|
|
|
|
|
|
Active site (Ca2+/ membrane binding)
|
5
|
3
|
3
|
3
|
3
|
100%
|
1 false positive prediction
|
|
HIV1-
reverse transcriptase
|
|
|
|
|
|
|
|
|
Catalytic site
|
5
|
5
|
3
|
5
|
3
|
60%
|
|
|
|
16
|
15
|
12
|
12
|
9
|
60%
|
Not a specific binding
|
|
SH3
domain
|
|
|
|
|
|
|
|
|
Peptide binding site
|
5
|
4
|
1
|
4
|
1
|
25%
|
|
|
Summary
|
58
|
50
|
34
|
44
|
28
|
56%
|
|
Table 1:
A summary of the 8 functional sites that
were analyzed in 5 domains and proteins. Columns from left to right: The second
column indicates the number of important residues that compose the functional
sites according to the literature. The third column: the number of exposed
residues out of the second column, as computed by the Surfv program. The forth
column: the number of exposed residues out of the second column, as predicted
by the neural net algorithm. The fifth column: the number of highly conserved
residues out of the third column as computed by the Rate4Site algorithm. The
sixth column (in yellow): the number of residues that are highly conserved and
predicted to be exposed out of the third column. The seventh column: the total
percentage success of the ConSeq server to predict the exposed annotated
functional residues as exposed and highly conserved.
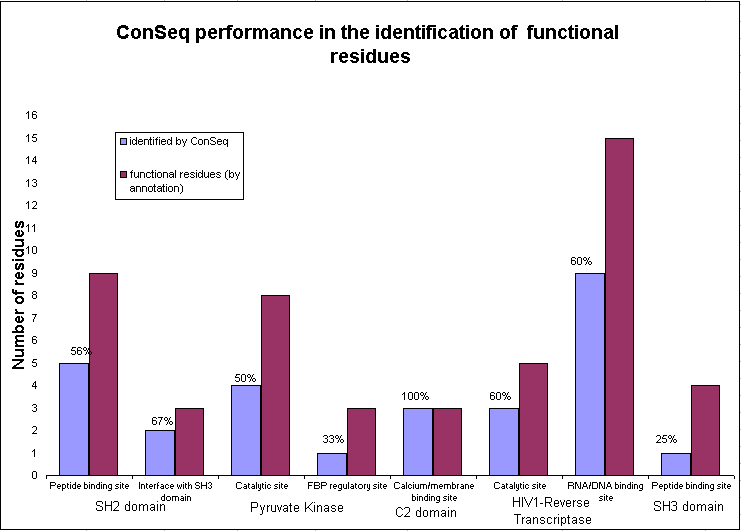
Figure 1: ConSeq's performance
(blue bars) in the identification of the exposed residues (Bordeaux bars) in 8
functional sites amongst 5 proteins
�
Example: The C2 domain
Click to see ConSeq result on-line: C2
domain
C2-domains are small, autonomously folded modules
of ~130 residues that are widely distributed. They are primarily found in
signal transduction, and in membrane-trafficking proteins such as phospholipases,
protein kinase C and synaptotagmins. C2-domains are involved in binding
phospholipids (membrane-docking) in a calcium-dependent or independent manner.
The C2 domain is composed of a scaffold of an eight-stranded, antiparallel β-sandwich that creates flexible loops on
the top and bottom of the domain. Many C2 domains bind calcium through a
cluster of 5 conserved aspartate residues in the upper loops region (Rizo and
Sudhof, 1998) (see Figure 2B). Calcium-independent C2 domains lack one
or more of the calcium-coordinating residues. Calcium binding increases the
electrostatic potential at the binding surface of the C2 domain, and increases
its attraction to acidic phopholipids. Nonspecific electrostatic interactions
have been shown to provide a major driving force for membrane association
in many C2 domains, including� the calcium-independent C2 domains. (Shao et
al., 1997; Murray and Honig, 2002).
We present here the
results obtained using an MSA of 50 closely homologous C2 domains
(synaptotagmins, protein kinase C, rabphilins etc.) collected from the
SWISS-PROT database using a profile generated with 5 PSI-BLAST iterations. We
analyzed the calcium-binding site of the first C2 domain of synaptotagmin 1
(C2A domain), which can bind 3 calcium ions (Figure 2B). All 5 aspartic residues are highly conserved according to Rate4Site. Three of the five Asp residues are exposed according to SurfV and we will therefore focus only on these three residues (Asp172, Asp232, Asp238). ConSeq results
for the C2 domain show that all 3 residues are highly conserved. However, 4
residues are predicted to be exposed, including one false positive (20%) hit in
Asp178. ConSeq identified the 3 exposed Asp residues as highly conserved
and exposed; therefore the success in this case is 100%, according to the
parameters we have set, i.e., to identify correctly only the exposed annotated residues (see Figures 2A and 2B below).
It is important to notice that the differentiation between the structural and
functional residues could be problematic for those that have both a
structural role and a functional role. For example, Asp230 binds two calcium
ions, and is therefore considered as a functional residue. However,it can also have a structural role in maintaining the active site conformation. Indeed this residue is buried within the protein core and therefore the
"structural" definition might be correct as well.
In addition to the active site residues, ConSeq identified some other functional residues that are involved in protein-protein interactions, for example His237 and Arg233, which take part in the interaction of the C2A domain with syntaxin (Shao et. al, 1997). The function of the other residues marked with "f" is still unknown, e.g. Gly175 that lies close to the calcium-binding site. Alternatively, they may be false positive exposure predictions; for example Thr195, that is known to have an important structural role, is predicted to be functional (Sutton et al., 1995). ConSeq also identified structurally important residues: Ala165, Leu168, Leu224 and Gly241. These residues are buried within the protein core; three are located in the �-strands region and one in the upper loops region, in close proximity to the active site.
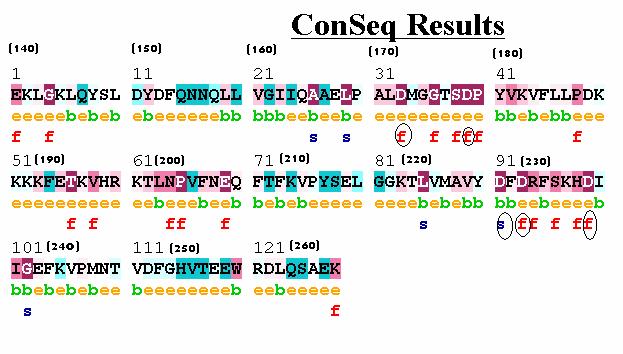


Figure 2: A. ConSeq results for the C2A domain of
synaptotagmin I. The five aspartate residues are circled. Four residues are
predicted to be exposed and Asp230 is predicted to have a structural role. Asp178
has a false positive surface accessibility prediction, i.e. it was wrongly predicted to be exposed.
B. Space-filling
model representation of the C2A domain from synaptotagmin I (PDB code: 1byn).
The conservation scores are color-coded onto each residue using the 9-grades
scale. The calcium-binding site is circled and calcium ions are in yellow. The
picture was obtained using the ConSurf server (http://consurf.tau.ac.il)
with the same MSA.
REFERENCES
1.
Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L.,
Eddy, S.R., Griffiths-Jones, S., Howe, K.L., Marshall, M. and Sonnhammer, E.L.
The Pfam Protein Families Database. Nucleic Acids Research 30(1): 276-280, 2002.
2.
Glaser, F., Pupko, T., Paz, I., Bell, R.E., Bechor, D., Martz, E.
and Ben-Tal, N. ConSurf: Identification of Functional Regions in Proteins by
Surface-Mapping of Phylogenetic Information. Bioinformatics 19:1-3, 2002.
3.
Mizuguchi, K., Deane, C.M., Blundell, T.L. and Overington, J.P.
HOMSTRAD: a database of protein structure alignments for homologous families. Protein
Sci. 7: 2469-2471,1998.
4.
Murray, D. and Honig,
B. Electrostatic control of the membrane targeting of C2 domains. Mol. Cell
9:145-154, 2002.
5.
Ren, J., Esnouf, R., Garman, E., Somers, D., Ross, C., Kirby, I.,
Keeling, J., Darby, G., Jones, Y., Stuart, D. and Stammers, D. High resolution structures of HIV-1 RT
from four RT-inhibitor complexes. Nat. Struct. Biol. 4: 293-302, 1995.
6.
Rizo, J. and Sudhof, T.C. Minireview: C2 domains, structure and
function of a universal Ca2+- binding domain. Jour. Biol. Chem., 273:
15879-15882, 1998.
7.
Shao, X., Li C., Fernandez, I., Zhang, X., Sudhof, T. C. and Rizo,
J. Synaptotagmin-Syntaxin interaction: the C2 domain as a Ca2+-dependent
electrostatic switch. Neuron 18: 133-142, 1997.
8.
Sridharan, S., Nicholls, A. and Honig, B. A new vertex algorithm to
calculate solvent accessible surface area. Biophysical Journal 61: A174, 1992.
9.
Sutton, R.B., Davletov, B.A., Berghuis, A. M., Sudhof, T. C. and
Sprang, S.R. Structure of the first C2 domain of Synaptotagmin I: a novel Ca2+/phospholipid-binding
fold. Cell 80: 929-938, 1995.
|