Comparison with other servers
We compared the ConSeq server results for eight active sites in 5 proteins with two other available sequence-based web servers: WebLogo server (Schneider and Stephens, 1990) and PROSITE (Falquet et al., 2002); the C2 domain example is presented in detail below.
Consensus sequence methods, e.g. CLUSTALW (Thompson et al., 1994), SEQUENCE LOGOS (Schneider et al., 1990) and motif searching tools (PROSITE; Bucher et al., 1994) are common methods to identify conserved residues amongst homologous sequences, and to provide insight into the function of the protein. However, these methods do not aim to distinguish between highly conserved residues with an important structural role and those with a functional role. The ConSeq server improves the above methods at three levels: first, the Rate4Site algorithm was shown to be a very sensitive and accurate method for estimating the rate of evolution of amino acid sites (inversely proportional to the degree of conservation) (Pupko et al., 2002). Secondly, the output consists of a buried or exposed-to-solvent prediction for each amino acid site, using a neural network-based method (Fariselli and Casadio, 2001). Thirdly, results are conveniently visualized.
WebLogo server: Sequence logos are a graphical representation of consensus sequences. Each logo consists of stacks of symbols, one stack for each position in the sequence. The overall height of the stack indicates the sequence conservation at that position, while the height of symbols within the stack indicates the relative frequency of each amino or nucleic acid at that position.
PROSITE is a database of protein families and domains. It is based on the observation that, while there is a huge number of different proteins, most of them can be grouped on the basis of similarities in their sequences into a limited number of families. By analyzing the constant and variable properties of such groups of similar sequences, it is possible to derive a signature for a protein family or domain, which distinguishes its members from all other unrelated proteins. These regions are generally important for the function of a protein and/or for the maintenance of its fold.
The calculations for the C2 domain with the WebLogo server were generated utilizing the same MSA of 50 homologous sequences used for the ConSeq server (see the Validation section and Figure 1, below). The results for the Weblogo server are presented in Figure 2.
Figure 1:
ConSeq results for the C2A domain of synaptotagmin I. The five aspartic acids (circled), which coordinate the 3 calcium ions, are highly conserved. Four residues are predicted to be solvent-exposed and Asp230 is predicted to have a structural role. Asp178 was wrongly predicted to be exposed.
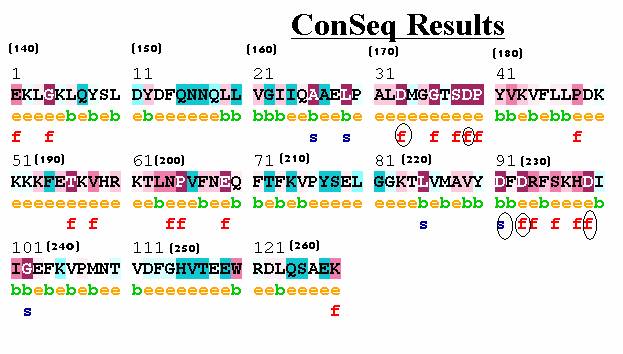

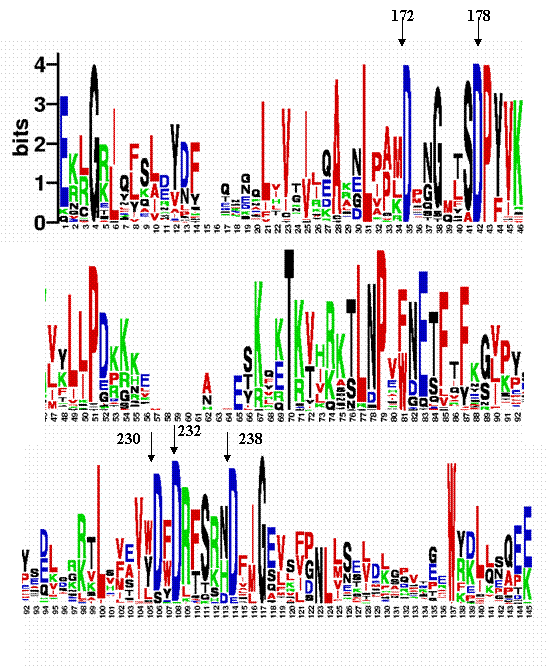
Figure 2:
Weblogo server results for C2A of synaptotagmin I. Arrows indicate the 5 functional aspartic acids. Asp172, Asp178 and Asp 232 are highly conserved, whereas Asp230 and Asp 238 are less conserved. WebLogo identifies the important structural residue, Thr195, as highly conserved. WebLogo does not distinguish between the important structural and functional residues. As we mentioned in the VALIDATION section, our definition of “structural” and “functional” residues might be problematic for the C2 domain active site because two Asp residues (Asp178, Asp230), which are buried, can be either functionally or structurally important in reality
We run PROSITE using the C2A domain of synaptotagmin I as a query sequence. PROSITE identified a C2 domain signature in the sequence. This signature contains only Asp 172 and Asp 178 as absolutely conserved residues, out of the 5 functional aspartic acids. Asp230, Asp232 and Asp238 are not part of the signature, even though these residues are important elements of the calcium-binding site (Figure 1B) (Rizo and Sudhof, 1998; Sutton et al., 1995) (Figure 3).
Figure 3: The PROSITE pattern that was detected for the sequence of the C2A domain of synaptotagmin I.
|
Description |
C2 domain signature. |
|
Pattern |
[ACG]-x(2)-L-x(2,3)-D-x(1,2)-[NGSTLIF]-[GTMR]-x-[STAP]-D-[PA]-[FY]. |
The Table below summarizes the comparison of ConSeq with the WebLogo server and PROSITE in identifying the annotated functional residues as highly conserved, in the eight active sites that were analyzed in the Validation section.
The ConSeq and the WebLogo servers were uploaded with the same MSA. The PROSITE server was scanned using the query sequence that was used for the ConSeq server.
The links to the results are highlighted.
|
Protein/Domain |
|
ConSeq |
WEBLOGO |
PROSITE |
|
Functional |
Highly conserved |
Highly conserved |
Highly conserved |
|
|
|
residues |
Residues |
Residues |
Residues |
|
|
(by annotation) |
(8+9 color grades) |
(~4 bits) |
(presented in a signature) |
|
SH2 domain |
|
|
|
|
|
Peptide binding site |
9 |
7 out of the 9 residues are highly conserved. |
6 out of the 9 residues are highly conserved. |
*profile |
|
SH3 interface |
3 |
2 of the 3 residues are highly conserved. |
1out of the 3 residues is highly conserved |
*profile |
|
Pyruvate Kinase |
|
|
|
|
|
Active site (pyruvate, K+, Mn2+, ATP binding) |
11 |
All 11 residues are highly conserved |
7 out of the 11 residues are highly conserved |
2 out of the 11 residues are highly conserved |
|
FBP binding site (allosteric regulation) |
4 |
All 4 residues are highly conserved. |
None of the residues is highly conserved. |
No pattern has been detected |
C2 domain |
|
|
|
|
|
Active site (Ca2+/ membrane binding) |
5 |
All 5 residues are highly conserved. |
3 out of the 5 residues are highly conserved. |
2 out of the 5 residues are highly conserved. |
|
HIV1- reverse transcriptase |
|
|
|
|
|
Catalytic site |
5 |
All 5 residues are highly conserved. |
2 out of the 5 residues are highly conserved. |
No pattern has been detected for the sequence |
RNA:DNA binding site |
16 |
12 of the 16 residues are highly conserved. |
5 out of the 16 residues are highly conserved. |
No pattern has been detected for the sequence |
|
SH3 domain |
|
|
|
|
|
Peptide binding site |
5 |
4 of the 5 residues are highly conserved. The fifth is color-graded 7. |
1 out of the 5 residues is highly conserved. |
*profile |
* PROSITE successfully identifies the sequences that were uploaded as SH2 and SH3 domains, using a profile. However, the profile does not discriminate between the highly conserved residues and the other residues in the domain; thus we could not compare these examples to the other servers.
Conclusion:
When comparing the ability of the three servers to identify the important functional residues as highly conserved, we found that ConSeq identifies most of the residues as highly conserved in the 8 functional sites. For example, in the active site of Pyruvate Kinase (PK), ConSeq identified all the 11 known residues as highly conserved, while Weblogo identified only 7 of them, and PROSITE successfully identified a motif that corresponds to the PK active site that highlights only 2 of the 11 residues.
Another advantage of the ConSeq server is the user-friendly visualization of the results. The results are easy to deduce and interpret, as compared to the results of the WebLogo server, for example.
REFERENCES
1. Falquet, L., Pagni, M., Bucher, P., Hulo, N., Sigrist, C.J, Hofmann, K. and Bairoch, A. The PROSITE database, its status in 2002. Nucleic Acids Res. 30, 235-238, 2002.
2. Glaser, F., Pupko, T., Paz, I., Bell, R.E., Bechor, D., Martz, E. and Ben-Tal, N. ConSurf: Identification of Functional Regions in Proteins by Surface-Mapping of Phylogenetic Information. Bioinformatics 19:1-3, 2002.
3. Pupko T., Bell R.E., Mayrose I., Glaser F. and Ben-Tal N. (2002). Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 18 Suppl. 1 S1-S7
4. Sridharan, S., Nicholls, A. and Honig, B. A new vertex algorithm to calculate solvent accessible surface area. Biophysical Journal 61: A174, 1992.
5. Thompson, J.D, Higgins, D.G. and Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice.Nucleic Acids Res. 22:4673-4680. 1994.